承認・届出等
販売名
ウィングスパン ステント
添付文書管理コード
22500BZX00505000_A_IP
承認番号
22500BZX00505000
承認・認証年月等
平成25年11月
一般的名称
一般的名称
70491000
脳動脈ステント
禁忌・禁止
(使用方法)
再使用禁止 (適応対象(患者)) ・本品留置後の抗血小板及び/又は抗凝固療法が禁忌である患者。[血栓症の発症リスクがより高まるおそれがある。]
|
形状・構造及び原理等
組成
ステント:ニッケル・チタン合金、プラチナ・イリジウム合金
デリバリーシステム:ナイロン、ステンレススチール、ポリエチレン
形状・構造及び原理等
本品は、自己拡張型ステントとデリバリーシステムからなるステントシステムである。デリバリーシステムはデリバリーカテーテル(アウターボディ及びインナーボディ)と回転式止血バルブから構成されており、また、自己拡張型ステントはあらかじめデリバリーカテーテル(アウターボディ)先端内腔に装填されている。
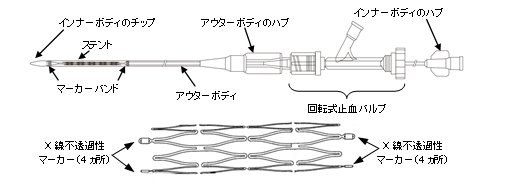
[原理]
本品ステントは、あらかじめステントデリバリーカテーテル先端部に装填されている。病変部においてカテーテルのアウターボディのハブを引き戻すことにより、ステントが自己拡張し、留置される。
使用目的又は効果
本品は、頭蓋内動脈狭窄症に対するバルーン拡張式血管形成術用カテーテルを用いた経皮的血管形成術において、以下の場合に使用する。
a.
血管形成術時に生じた血管解離、急性閉塞又は切迫閉塞に対する緊急処置
b.
他に有効な治療法がないと判断される血管形成術後の再治療
使用方法等
使用方法
<ステントシステムの準備>
<ガイドワイヤの位置決定>
<ステント位置決定および拡張>
<血管造影法による病変とステントの選択>
★1:病変の両側で最低3mm確保できるよう、病変より6mm長いステント長を選択する。
★2:展開後ステント径は最小値を示す。
★3:この表で推奨されたサイズと、大きい方の血管径(血管直径の近位または遠位)の両方を元に、ステント径を選択する。
(1)
血管造影を使用して、病変の位置と範囲および血管径を測定する。ステント留置の成功には慎重なステントのサイズ選択が重要となる。一般的に、ステントサイズは病変に隣接する正常な血管径に適合するように選択する。
(2)
病変の両側で最低3mm確保できるよう、病変より少なくとも6mm長いステント長を選択する。各ステントの直径に対応する最大ステントサイズのガイドラインを表1に示す。
| 表1 推奨サイズガイドライン | ||||||
| 表示 ステント径 (mm) | 表示 ステント長1 (mm) | 展開後 ステント径2 (mm) | 推奨 血管径3 (mm) | 有効長 (cm) | 最大 ガイド ワイヤ径 | 最小 ガイディング カテーテル 内径 |
| 2.5 | 9 | 2.8 | 2.0超、 2.5以下 | 135 | 0.36mm (0.014in) | 1.63mm (0.064in) |
| 15 | ||||||
| 20 | ||||||
| 3.0 | 9 | 3.4 | 2.5超、 3.0以下 |
|||
| 15 | ||||||
| 20 | ||||||
| 3.5 | 9 | 3.9 | 3.0超、 3.5以下 |
|||
| 15 | ||||||
| 20 | ||||||
| 4.0 | 9 | 4.4 | 3.5超、 4.0以下 |
|||
| 15 | ||||||
| 20 | ||||||
| 4.5 | 9 | 4.9 | 4.0超、 4.5以下 |
|||
| 15 | ||||||
| 20 | ||||||
★2:展開後ステント径は最小値を示す。
★3:この表で推奨されたサイズと、大きい方の血管径(血管直径の近位または遠位)の両方を元に、ステント径を選択する。
<ステントシステムの準備>
(1)
滅菌パウチを開封し、包装トレイを取り出し、包装に損傷がないか確認する。
(2)
ディスペンサーフープをヘパリン加生理食塩液でフラッシュし、親水性コーティングを湿潤させ、トレイから一体化されたハブおよび回転式止血バルブを慎重に取り出し、インナーボディが動かないように回転式止血バルブを締め、デリバリーシステム全体をフープから取り出す。デリバリーシステムにキンクなどの損傷がないか確認する。また、ステントがデリバリーシステムの先端部に装填されていることを確認する。
注意:アウターボディ手元部の回転式止血バルブは過度な力をかけて締めつけないこと。
(3)
回転式止血バルブをデリバリーシステムのインナーボディのハブに取り付け、インナーボディのガイドワイヤルーメンをヘパリン加生理食塩液でフラッシュする。
(4)
アウターボディの回転式止血バルブを緩め、ヘパリン加生理食塩液でアウターボディをフラッシュし、インナーボディの上で回転式止血バルブを締める。
(5)
システムから空気を除去するために、アウターボディをフラッシュし続ける。
(6)
加圧したヘパリン加生理食塩液でフラッシュするため、アウターボディの回転式止血バルブのサイドポートと、インナーボディに接続した回転式止血バルブのサイドポートに、ヘパリン加生理食塩液の持続的輸液ラインを接続する。
(7)
インナーボディの上に固定されたアウターボディの止血バルブを緩め、インナーボディのデュアルテーパーチップの近位端とアウターボディの遠位端の間の隙間が1〜2mmになるように、インナーボディを押さえながらアウターボディをゆっくりと引き戻す。これによりアウターボディの先端から生理食塩液が滴下する。
注意:過剰な力を加えたり、インナーボディのチップをアウターボディの中に強く引き込み過ぎないように注意すること。
(8)
ステントシステムを前進させている間は、インナーボディを定位置に固定するため、アウターボディの回転式止血バルブを締め、インナーボディを固定する。
<ガイドワイヤの位置決定>
(1)
準的なマイクロカテーテルとガイドワイヤ技術で、病変に沿ってアクセスガイドワイヤの位置を決定すること。推奨するガイディングカテーテルの仕様は、90cm長以上、最小内径は1.63mm (0.064in)である。
(2)
アクセスガイドワイヤを0.36mm (0.014in)のエクスチェンジガイドワイヤに取り替え、マイクロカテーテルを引き出す。病変に沿ってエクスチェンジガイドワイヤは残しておく。スティッフタイプガイドワイヤよりもソフトタイプガイドワイヤが推奨される。
<ステント位置決定および拡張>
(1)
0.36mm (0.014in)ガイドワイヤ上にステントシステムを慎重に後ろから導入する。
(2)
ステントシステムをガイディングカテーテル内に慎重に進める。
(3)
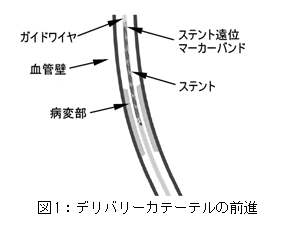
X線透視下で、ステントが標的病変部のやや遠位にくるまでステントシステムを前進させる(ステントの位置は、4つの遠位X線不透過性マーカーで確認する)。
図1:デリバリーカテーテルの前進を参照
図1:デリバリーカテーテルの前進を参照
(4)
アウターボディの回転式止血バルブを緩め、インナーボディのX線不透過性の近位バンパーがステント近位端のマーカーに達するまでインナーボディを前進させる。その後アウターボディの回転式止血バルブを締める。
(5)
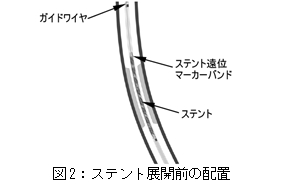
ステントが標的病変部に合う位置までアウターボディのハブを引き戻し、ステントの最終的な位置決めを行う。デリバリーシステムをわずかに引き戻してステントの位置の最終調整をすることにより、ステント展開前にデリバリーシステムのたわみを取り除くことができる。
図2:ステント展開前の配置を参照。
図2:ステント展開前の配置を参照。
(6)
ステント展開の準備完了。
注意:アウターボディ回転式止血バルブは過度な力をかけて締めつけないこと。
(7)
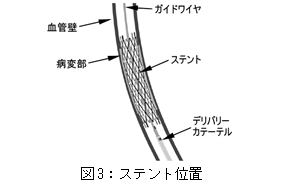
アウターボディの回転式止血バルブを緩める。片手でインナーボディのハブを固定し、もう片方の手で慎重にアウターボディのハブを引き戻してステントを展開する。
図3:ステント位置を参照。
図3:ステント位置を参照。
(8)
ステントが展開すると、ステント遠位端のX線不透過性マーカーが広がっていくことが分かる。連続的にスムースな操作でステントを展開し続ける。ステントを展開し始めたら、決してステント位置は移動させないこと。ステントが展開している時、アウターボディを前進させないように注意すること。
(9)
ステントが完全に展開した後、アウターボディの回転式止血バルブを締め、慎重にデリバリーシステムを抜去する。システムを抜去する際、過度の摩擦を感じた場合には、アウターボディの回転式止血バルブを緩め、アウターボディの先端がインナーボディのチップに接触するまでアウターボディを進める。回転式止血バルブを締めてデリバリーシステムを抜去する。
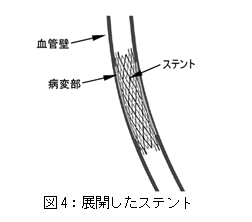
図4:展開したステントを参照。
図4:展開したステントを参照。
使用上の注意
1.重要な基本的注意
<一般的な注意>
<準備上の注意>
<手技中の注意>
<MRIに関する情報>
(1)
金属に対する過敏症が明らかな患者では、本品の留置によりアレルギー反応が生じるおそれがあるので使用しないこと。[本品は血管内に留置して使用されるものであり、含有金属が溶出することにより金属アレルギーを惹起するおそれがある。このような場合にはステント本来の効果が減弱するおそれがあるとする報告があるので、必ず問診を行い、金属アレルギーの患者については本品を用いた治療を実施することの妥当性について再検討を行うこと。]
※参考文献「Lancet 2000; 12: 1895-1897」
※参考文献「Lancet 2000; 12: 1895-1897」
(2)
本品の適切な選択は患者の安全を確保する上で重要である。術前に血管造影を行い、対象部位の評価を十分に行うこと。
(3)
一般的にステント留置術は狭窄リスクを伴うことが報告されている。同様に本品を留置した血管部位についても狭窄が発現した場合は狭窄部位に対する拡張術が必要になることがあるが、本品を留置した血管部位の拡張後のリスク及び長期転帰については現時点では確立されていない。
(4)
本品のステントは、病変の両側を3mm以上カバーするステントのサイズ(長さ)を選択すること。
(5)
本品は、造影剤またはヘパリン加生理食塩液以外を注入することを設計または意図していない。
(6)
動物実験で見られた血管狭窄や新生内膜の肥厚は、ステントの留置または放射状拡張による血管壁外傷が生じることに関連していると考えられている。
(7)
ステントまたは塞栓コイルのような、他に埋め込まれた金属と隣接または接触して留置する場合には、金属が電解腐食することがないように類似原材料を使用すること。
(8)
ラベルの表示が不完全であったり、判読できない場合は、本品を使用しないこと。
(9)
一般的にステント留置術は狭窄リスクを伴うことが報告されている。同様に本品を留置した血管部位についても狭窄が発現した場合は狭窄部位に対する拡張術が必要になることがあるが、本品を留置した血管部位の拡張後のリスク及び長期転帰については現時点では確立されていない。[予測できない有害事象が発現するおそれがある。]
<準備上の注意>
(1)
使用前に滅菌パウチ及び本品を慎重に点検し、輸送中に損傷していないことを確認すること。部品がキンク(折れ)していたり、損傷している場合は使用しないこと。[本品の性能が十分に発揮できない。または感染症を引き起こすおそれがある。]
(2)
非侵襲的頭蓋内手技で使用する規定の抗血小板/抗凝固療法はステント治療において重要である。ステント留置後、医師は患者に処方を守るよう指導し、内科治療で対応しないリスクについて助言すること。適切な抗血小板/抗凝固剤療法で管理されない場合には、手技中にステント留置による血栓症が起こるおそれがある。[血栓塞栓性事象の発症リスクを高めるおそれがある。]
(3)
ステントまたはデリバリーシステムに損傷が生じるおそれがあるため、ステントシステムの先端を蒸気にあてて成形しないこと。
<手技中の注意>
(1)
ステントを埋植することにより、ステントの遠位または近位の血管解離が生じたり、更なる非侵襲的治療(すなわち、更なる血管拡張や、ステントの留置)が必要となるような他の合併症(血管攣縮/急性血管閉塞)の原因となることがある。
(2)
血管内でステントの位置決めが適切にできない場合には、ステントを展開しないこと。
(3)
本品の操作中に過度の抵抗が感じられた場合、手技のどの段階であっても使用を中止すること。抵抗に逆らって操作すると、血管又は本品の構成品を損傷する可能性がある。
(4)
本品を用いた手技は高画質の透視観察下で行うこと。[本品及び併用機器を視認できず、血管が損傷するおそれがある。]
(5)
ステントを留置することで、血管の側枝の開存性が保てなくなることがある。
(6)
ステントの位置変更または抜去のために本品を使用しないこと。
(7)
併用する機器を、留置した本品ステント内腔を通過させる場合には、十分注意すること。[併用機器等の損傷あるいは本品ステントが移動するおそれがある。]
(8)
蛇行性の血管内では、スティッフタイプガイドワイヤは手技中にステントシステムの中で動かなくなるおそれがある。そのような場合にはソフトタイプガイドワイヤのみを使用すること。
(9)
留置後、本品ステントは表示径2.5mmのステントでは2.4%まで、表示径4.5mmのステントでは7.1%まで短縮することがある。
(10)
ステント除去術(ワイヤ、スネア、鉗子の追加使用)では、血管系または血管系の接近した側に、結果として更なる外傷を生じることがある。合併症には、出血、血腫または偽動脈瘤が含まれる。
<MRIに関する情報>
非臨床試験の結果、本品は単独で留置した場合、あるいは2本目のステントとオーバーラップさせた場合、以下の条件で留置直後に安全なスキャンが可能である。
(1)
静磁場強度が1.5又は3テスラ以下
(2)
空間勾配磁場が2,500ガウス/cm(25テスラ/m)以下
(3)
通常操作モードの勾配およびSAR(2.0W/kg 未満の最大全身平均比吸収率(SAR)および3.2W/kg 未満の最大頭部SAR)での、スキャンシーケンス毎に15 分以下(RF 曝露を伴う)の合計MR スキャン時間。
非臨床試験を解析したところ、本品の生体内での温度上昇は、1.5 テスラおよび3テスラのMR システムでの通常操作モードにおいて、15 分間のMR スキャン後、4℃以下であった。本品なこのMRI環境において移動はしない。
関心領域が本品留置部位、もしくはその周辺である場合、MR 画像の質が低下することがある。そのため、MR 画像のパラメータを本品に合わせることが必要な場合がある。
関心領域が本品留置部位、もしくはその周辺である場合、MR 画像の質が低下することがある。そのため、MR 画像のパラメータを本品に合わせることが必要な場合がある。
2.不具合
本品の使用によって、以下のような不具合が起こり得るが、これらに限定されるものではない。
重大な不具合
(1)
ステントの移動
(2)
ステントの誤留置
(3)
ステントの破損
(4)
ステントデリバリー時、または展開時の摩擦
3.有害事象
本品の使用によって、以下のような有害事象が起こり得るが、これらに限定されるものではない。
重大な有害事象
(1)
アレルギー反応
(2)
動脈瘤
(3)
脳虚血
(4)
凝血障害
(5)
死亡
(6)
塞栓(空気、組織、血栓による)
(7)
出血
(8)
循環血流量過多
(9)
感染症
(10)
ステント内再狭窄
(11)
虚血/梗塞
(12)
神経学的症状
(13)
再狭窄
(14)
疼痛
(15)
偽動脈瘤
(16)
発作
(17)
ステント閉塞
(18)
ステント塞栓
(19)
ステント血栓症
(20)
脳卒中
(21)
失神
(22)
一過性脳虚血発作(TIA)
(23)
血管攣縮
(24)
血管解離
(25)
血管閉塞
(26)
血管穿孔
(27)
血管破裂
(28)
血管痙攣
(29)
血管血栓症
(30)
外科的修復術またはインターベンションを必要とする血管の外傷
その他の有害事象
(1)
造影剤/抗血小板薬に対するアレルギー反応
(2)
血腫、疼痛および/または挿入部位の感染
(3)
高血圧/低血圧
臨床成績
1.
国内臨床試験
国内臨床試験は薬物療法に抵抗性を示し、本品が到達可能な狭窄度50%以上の頭蓋内動脈狭窄に起因する一過性脳虚血発作又は脳卒中患者20例が登録された。本器留置前に重篤な有害事象が発現した1例を除き、19例に本器による手技を行った。本試験の主要評価項目である「手技6ヵ月後までの同側脳卒中又は死亡の発生」の割合は19例中2例であった。
本試験中に発現したすべての有害事象は19例(95.0%)87件であった。
治験機器又は手技との因果関係が否定できない有害事象は12例(60.0%)21件に発現した。そのうち、治験機器との因果関係が否定できない有害事象は5例(25.0%)6件に認められた。
本試験中に発現したすべての有害事象は19例(95.0%)87件であった。
治験機器又は手技との因果関係が否定できない有害事象は12例(60.0%)21件に発現した。そのうち、治験機器との因果関係が否定できない有害事象は5例(25.0%)6件に認められた。
| [国内臨床試験で実施された抗血小板療法] <手技前(手技3日以上前から手技当日まで)> 下記の薬物のうち2剤以上の併用療法を行うことが推奨された。 アスピリン(81〜324mg/日)、クロピドグレル(75mg/日)、シロスタゾール(100〜200mg/日)又はチクロピジン(100〜200mg/日)。 <手技後(手技の翌日から)> 上述の抗血小板療法を手技の翌日から少なくとも4週間実施することが推奨された。その後医師の判断により薬剤の種類と量を変更できるが、少なくとも1剤の抗血小板薬を無期限に投与を継続することが推奨された。 |
注)併用する抗血小板薬の添付文書を必ず参照すること。なお、チクロピジン塩酸塩製剤の投与においては、血栓性血小板減少性紫斑病(TTP)、無顆粒球症、重篤な肝障害等の重大な副作用が、主に投与開始後2ヶ月以内に発現し、死亡に至る例も報告されているので、投与開始後2ヶ月間は、原則として1回2週間分を処方するとともに、以下の点に十分留意すること。また、クロピドグレル硫酸塩製剤を投与する場合においても、同様に以下の点に留意すること。
(1)
投与開始後2ヶ月間は、特に上記の副作用の初期症状の発現に十分留意し、原則として2週間に1回、血球算定(白血球分画を含む)、肝機能検査を行い、上記副作用の発現が認められた場合には、投与を中止し、適切な処置を行うこと。本剤投与期間中は、定期的に血液検査を行い、上記副作用の発現に注意すること。
(2)
本剤投与中、患者の状態から血栓性血小板減少性紫斑病、顆粒球減少、肝障害の発現等が疑われた場合には、必要に応じて血液像もしくは肝機能検査を実施し、適切な処置を行うこと。
2.
海外臨床試験
[Wingspan and Gateway Safety Study]
本試験は頭蓋内アテローム性疾患に起因する再発性脳卒中を有し、薬物療法に抵抗性を示し、本品が到達可能な狭窄度50%以上の頭蓋内動脈狭窄を有する患者45例が登録された。本品留置前にPTAバルーンカテーテルが病巣へアクセスできなかった1例を除き44例に本機器による手技を行った。本試験の主要安全性評価項目である「手技30日後の同側脳卒中又は死亡」は4.5%であった。主要有効性評価項目は「ステント留置成功」及び「手技成功」とされ、それぞれ100%、97.7%であった。
本試験中に発現した重篤な有害事象は18例(40.0%)26件であった。このうち治験機器又は手技との因果関係が否定できない重篤な有害事象は8件に認められた。
本試験中に発現した重篤な有害事象は18例(40.0%)26件であった。このうち治験機器又は手技との因果関係が否定できない重篤な有害事象は8件に認められた。
| [Wingspan and Gateway Safety Studyで実施された抗血小板療法] <手技前> 手技3日前からクロピドグレル(75mg)、アスピリン(300mg又は325mg)を毎日投与、又はクロピドグレル(225mg)、アスピリン(300〜650mg)を手技前日に投与することが推奨された。 <手技後> クロピドグレル(75mg)を30日間投与、アスピリン(300mg又は325mg)を無期限に投与することが推奨された。 |
[SAMMPRIS試験]
本試験は頭蓋内主幹動脈の70%以上の狭窄病変を有し、一過性脳虚血発作又は脳梗塞を30日以内に発症した患者を対象に、積極的内科治療の成績と積極的内科治療に加え経皮的バルーン血管形成術後に本品を用いたステント留置術(PTAS)を行った際の治療成績を比較する多施設無作為化比較試験である。主要評価項目は「登録後又は標的血管の再灌流手技後30日以内の脳卒中及び死亡」及び「30日以降の標的血管領域に起きた脳梗塞」とされた。
451例登録(積極的内科治療群227例、PTAS群224例)の時点で「30日以内の脳卒中及び死亡」が積極的内科治療群で5.8%、PTAS群で14.7%(p=0.002)、「1年以内の標的血管領域の脳卒中及び死亡が積極的内科治療群で12.2%、PTAS群で20.0%(p=0.009)との結果が出たため、本試験への患者登録が打ち切られた。
451例登録(積極的内科治療群227例、PTAS群224例)の時点で「30日以内の脳卒中及び死亡」が積極的内科治療群で5.8%、PTAS群で14.7%(p=0.002)、「1年以内の標的血管領域の脳卒中及び死亡が積極的内科治療群で12.2%、PTAS群で20.0%(p=0.009)との結果が出たため、本試験への患者登録が打ち切られた。
| 積極的内科治療群 (n=227) | PTAS群 (n=224) | P値 | ||
| 30日以内の脳卒中及び死亡 | 5.8% | 14.7% | 0.002 | |
| 30日以降の標的血管領域に起きた脳梗塞 | 5.7% | 5.8% | − | |
| 1年以内の脳卒中及び死亡 | 12.2% | 20.0% | 0.009 | |
| 全脳卒中又は死亡 | 17.5% | 23.4% | 0.06 | |
| 全脳卒中 | 14.9% | 22.3% | 0.03 | |
| 死亡 | 4.1% | 3.4% | 0.95 | |
| 致死性の脳卒中 | 6.4% | 9.0% | 0.21 | |
| 心筋梗塞 | 4.0% | 2.2% | 0.60 | |
| 脳卒中に関連しない出血性合併症 | 1.4% | 3.6% | 0.10 | |
| 全出血性合併症 | 1.8% | 9.0% | <0.001 | |
| [SAMMPRIS試験で実施された抗血小板療法 PTAS群] 組み入れ後からアスピリン(325mg/日)をフォローアップまで継続。クロピドグレル(75mg/日)を90日間継続。 クロピドグレル(75mg/日)はPTASの5日前から、また600mgをPTASの6時間と24時間前に服用。 |
なお本試験は米国NIHファンドによって実施された臨床試験である。
※参考文献「N Engl J Med 2011; Sep 15;365(11):993-1003」
保管方法及び有効期間等
保管方法**
**高温、多湿、直射日光をさけ室温で保管
有効期間**
**有効期間:外箱の表示を参照(自己認証による)
承認条件
1.
頭蓋内動脈狭窄症の治療に関する十分な知識・経験を有する医師により、同治療に伴う合併症への対応ができる体制が整った医療機関において、本品が使用されるよう、関連学会と連携の上で必要な措置を講ずること。
2.
1に掲げる医師が、適応を遵守し、講習の受講等により、本品を用いた血管内治療に関する手技及び同治療に伴う合併症等に関する十分な知識を得た上で、本品が用いられるよう、関連学会と連携の上で必要な措置を講ずること。
3.
一定数の症例が集積されるまでの間は、本品を使用する症例全例を対象として、使用成績調査を行い、経年解析結果を医薬品医療機器総合機構宛て報告するとともに、必要に応じ適切な措置を講ずること。
製造販売業者及び製造業者の氏名又は名称等
氏名又は名称(製造販売業の種別)
日本ストライカー株式会社
第一種医療機器製造販売業
住所等
電話番号
03-6894-0000(代表)