ベージニオ錠50mg/ベージニオ錠100mg/ベージニオ錠150mg
作成又は改訂年月
| * | 2021年 12月改訂 ( 第2版 、 効能変更、用法変更 ) |
| 2020年 4月改訂 ( 第1版 ) |
日本標準商品分類番号
薬効分類名
承認等
ベージニオ錠50mg
販売名コード
YJコード
販売名英語表記
承認番号等
承認番号
販売開始年月
貯法・有効期間
貯法
有効期間
規制区分
劇薬
処方箋医薬品 注)
注) 注意―医師等の処方箋により使用することベージニオ錠100mg
販売名コード
YJコード
販売名英語表記
承認番号等
承認番号
販売開始年月
貯法・有効期間
貯法
有効期間
規制区分
劇薬
処方箋医薬品 注)
注) 注意―医師等の処方箋により使用することベージニオ錠150mg
販売名コード
YJコード
販売名英語表記
承認番号等
承認番号
販売開始年月
貯法・有効期間
貯法
有効期間
規制区分
劇薬
処方箋医薬品 注)
注) 注意―医師等の処方箋により使用すること一般的名称
1. 警告
- 1.1 *本剤は、緊急時に十分対応できる医療施設において、がん化学療法に十分な知識・経験を持つ医師のもとで、本剤の投与が適切と判断される症例についてのみ投与すること。また、治療開始に先立ち、患者又はその家族に有効性及び危険性を十分説明し、同意を得てから投与すること。
- 1.2 *本剤投与開始前に、胸部CT等の検査及び問診を実施し、間質性肺疾患の合併又は既往歴の有無を確認した上で、投与の可否を慎重に判断すること。,
- 1.3 *間質性肺疾患があらわれ、死亡に至った症例も報告されているので、初期症状(呼吸困難、咳嗽、発熱等)の確認、動脈血酸素飽和度(SpO2)の検査及び胸部X線検査の実施等、患者の状態を十分に観察すること。異常が認められた場合には、速やかに本剤を休薬し、呼吸器疾患に精通した医師と連携の上、胸部CT等の検査を実施するとともに、適切な処置を行うこと。本剤による間質性肺疾患と診断された場合は、本剤の投与を中止すること。,,,
5. 効能又は効果に関連する注意
6. 用法及び用量
*内分泌療法剤との併用において、通常、成人にはアベマシクリブとして1回150mgを1日2回経口投与する。ただし、術後薬物療法の場合には、投与期間は24ヵ月間までとする。なお、患者の状態により適宜減量する。
7. 用法及び用量に関連する注意
- 7.1 *併用する内分泌療法剤等について、「17. 臨床成績」の項の内容を熟知し、本剤の有効性及び安全性を十分に理解した上で、選択を行うこと。,,
- 7.2 *副作用があらわれた場合は、以下の基準を考慮して、休薬・減量・中止すること。
- 副作用発現時の用量調節基準
副作用
程度 注1)
処置
肝機能障害
持続する又は再発のグレード2のAST又はALT増加
ベースライン又はグレード1以下に回復するまで休薬する。
再開する場合には投与量を1段階減量する。
グレード3のAST又はALT増加
グレード2以上のAST又はALT増加、かつ総ビリルビンが基準値上限の2倍超 注2)
投与を中止する。
グレード4のAST又はALT増加
下痢
グレード2で24時間以内に回復しない場合
グレード1以下に回復するまで休薬する。
再開する場合には減量は不要である。
治療しても症状が継続する又は減量せずに再開後に再発したグレード2
グレード1以下に回復するまで休薬する。
再開する場合には投与量を1段階減量する。
入院を要する又はグレード3もしくは4
血液毒性
グレード3(初回発現)
グレード2以下に回復するまで休薬する。
再開する場合には必要に応じて投与量を1段階減量する。
グレード3(2回目以降の発現)又は4
グレード2以下に回復するまで休薬する。
再開する場合には投与量を1段階減量する。
G-CSF製剤を投与した場合
G-CSF製剤の最終投与後少なくとも48時間以上経過し、かつグレード2以下になるまで休薬する。
再開する場合には投与量を1段階減量する。
投与を中止する。
静脈血栓塞栓症
(術後薬物療法と
しての投与時)
グレード2~4
投与を中止する、又は適切な治療を行い、状態が安定するまで休薬する。再開する場合には必要に応じて投与量を1段階減量する。
上記以外の副作用
治療しても症状が継続する又は再発のグレード2で、7日以内にベースライン又はグレード1まで回復しない場合
ベースライン又はグレード1以下に回復するまで必要に応じて休薬する。
再開する場合には必要に応じて投与量を1段階減量する。
グレード3又は4
ベースライン又はグレード1以下に回復するまで休薬する。
再開する場合には投与量を1段階減量する。
注1) グレードはNCI-CTCAE ver. 4.0に準じる。注2) 明らかな胆汁うっ滞を認めない場合
8. 重要な基本的注意
9. 特定の背景を有する患者に関する注意
10. 相互作用
10.2 併用注意(併用に注意すること)
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
|
本剤の血中濃度が上昇するおそれがあるので、減量を考慮するとともに、患者の状態を慎重に観察し、有害事象の発現に十分注意すること。 |
これらの薬剤等がCYP3Aの代謝活性を阻害するため、本剤の血中濃度を上昇させる可能性がある。 |
|
|
グレープフルーツ |
本剤服用時は飲食を避けること。 |
これらの薬剤等がCYP3Aの代謝活性を阻害するため、本剤の血中濃度を上昇させる可能性がある。 |
|
本剤の血中濃度が低下し、効果が減弱するおそれがあるので、CYP3A誘導作用のない薬剤への代替を考慮すること。 |
これらの薬剤がCYP3Aの代謝酵素を誘導するため、本剤の血中濃度を低下させる可能性がある。 |
11. 副作用
11.1 重大な副作用
- 11.1.1 *肝機能障害
- 11.1.2 *重度の下痢(7.5% 注3) )
-
11.1.3 *骨髄抑制
好中球減少(42.2%)、白血球減少(32.0%)、貧血(19.1%)、血小板減少(11.0%)、リンパ球減少(10.6%)等があらわれることがある。
-
11.1.4 *間質性肺疾患(1.2%)
異常が認められた場合には、休薬し、呼吸器疾患に精通した医師と連携の上、胸部CT等の検査を実施するとともに、適切な処置を行うこと。,,,
-
11.1.5 *静脈血栓塞栓症(1.7%)
深部静脈血栓症(0.7%)、肺塞栓症(0.6%)等の静脈血栓塞栓症があらわれることがある。
注3) NCI-CTCAE ver. 4.0のグレード3以上の副作用
11.2 その他の副作用
|
20%以上 |
5~20%未満 |
5%未満 |
|
|---|---|---|---|
|
*消化器 |
下痢、腹痛、悪心 |
嘔吐、食欲減退、口内炎 |
消化不良、便秘、胃炎 |
|
呼吸器 |
咳嗽、呼吸困難 |
||
|
*循環器 |
高血圧 |
||
|
*感染 |
上気道感染、尿路感染、肺感染、上咽頭炎、結膜炎、副鼻腔炎、膣感染、咽頭炎、敗血症 |
||
|
*皮膚 |
脱毛症、発疹 |
そう痒症、皮膚乾燥、爪の障害、ざ瘡様皮膚炎 |
|
|
*精神神経系 |
浮動性めまい、味覚異常、うつ病 |
||
|
*臨床検査値異常 |
血中クレアチニン増加 |
低カリウム血症、γ-GTP増加、高カリウム血症、低カルシウム血症 |
|
|
*その他 |
疲労 |
ほてり、頭痛 |
流涙増加、体重減少、倦怠感、発熱、四肢痛、末梢性浮腫、眼乾燥、脱水、筋力低下、インフルエンザ様疾患、悪寒 |
14. 適用上の注意
16. 薬物動態
16.1 血中濃度
-
16.1.1 単回投与
日本人進行癌患者12例に本剤100、150及び200mg 注10) を単回経口投与したときの血漿中濃度推移及び薬物動態パラメータは以下のとおりであった1) 。

図1)本剤100、150及び200mg 注10) を単回経口投与したときの血漿中濃度推移(平均値) 表1)本剤100、150及び200mg 注10) を単回経口投与したときの薬物動態パラメータ
(幾何平均値及び変動係数%)100mg 注10)
150mg
200mg 注10)
例数
3
3
6
Cmax
(ng/mL)127
(51)167
(40)214
(87)tmax 注4) (hr)
5.93
(5.92-7.98)5.95
(3.95-6.05)4.97
(3.95-5.95)AUC0-∞
(ng・hr/mL)6970,6450 注5)
4450
(39)5480
(95)CL/F
(L/hr)14.3,15.5 注5)
33.7
(39)36.5
(95)VSS/F
(L)637,577 注5)
1120
(41)947
(90)t1/2 注6) (hr)
27.5,24.1 注5)
21.9
(19.3-24.6)16.3
(14.2-22.6)注4) 中央値(範囲)注5) 個別値(例数=2)注6) 幾何平均値(範囲) -
16.1.2 反復投与
進行癌患者116例に本剤100、150及び200mg 注10) を1日2回反復経口投与したときの血漿中濃度推移及び薬物動態パラメータは以下のとおりであった。血漿中濃度は反復投与後5日に定常状態に到達した2) (外国人データ)。
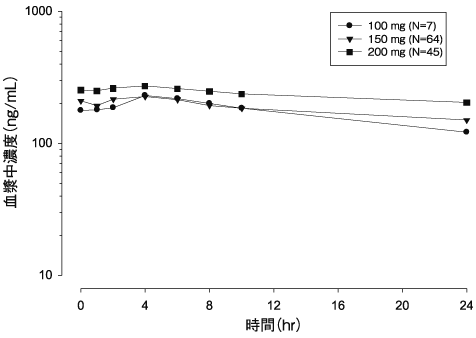
図2)本剤100、150及び200mg 注10) を1日2回反復経口投与後(第1サイクル第28日目)の血漿中濃度推移(平均値) 表2)本剤100、150及び200mg 注10) を1日2回反復経口投与後(第1サイクル第28日目)の薬物動態パラメータ(幾何平均値及び変動係数%) 100mg 注10) 1日2回
150mg 1日2回
200mg 注10) 1日2回
例数
7
64
45
Cmax,ss
(ng/mL)240
(52)
251
(89)
305
(77)tmax,ss 注7) (hr)
4.00
(2.00-6.03)
3.97
(0.00-10.15)
4.08
(0.00-10.00)AUCτ,ss
(ng・hr/mL)2400
(54)
2380 注8)
(95)
3120 注9)
(72)
注7) 中央値(範囲)注8) 例数=63注9) 例数=43τ:投与間隔(12時間)
16.2 吸収
16.3 分布
アベマシクリブのヒト血漿蛋白結合率は高く(平均値:約96~98%)、152~5066ng/mLまでの濃度範囲では濃度依存性は見られなかった。アベマシクリブは、血清アルブミン及びα1-酸性糖蛋白質と結合する(in vitro)5) 。
アベマシクリブと同程度の活性を有する主要代謝物であるM2、水酸化N-脱エチル体(M18)及び水酸化体(M20)のヒト血漿蛋白結合率も高く、約89~94%であった。
16.4 代謝
16.5 排泄
アベマシクリブは主に肝代謝により消失する。健康成人6例に[14C]-アベマシクリブ150mgを単回経口投与後336時間までに、投与量の約81%が糞便中に排泄され、約3.4%が尿中に排泄された。糞便中に検出された放射能のほとんどは代謝物であった7) (外国人データ)。
16.6 特定の背景を有する患者
16.7 薬物相互作用
17. 臨床成績
17.1 有効性及び安全性に関する試験
-
17.1.1 国際共同第III相無作為化比較試験(MONARCH2試験)
ホルモン受容体陽性かつHER2陰性であり、内分泌療法歴のある手術不能又は再発乳癌患者669例を対象に、本剤+フルベストラントとプラセボ+フルベストラントを比較する無作為化二重盲検プラセボ対照第III相国際共同試験を実施した。
本剤150mg 注11) 又はプラセボ(1日2回)とフルベストラント500mg(1サイクルを28日間として、第1サイクルの1及び15日目並びに第2サイクル以降の1日目)を病態の悪化等が認められるまで投与を継続した。
主要評価項目である治験責任医師判定による無増悪生存期間について、本剤+フルベストラントの併用投与により、プラセボ+フルベストラントの併用投与と比較して統計学的に有意な延長が認められた12) 。
注11) 試験開始時には本剤の用量は200mgと設定されたものの、下痢等により休薬又は減量に至った症例が多数認められたことから、本剤の投与を新たに開始する患者及び200mgで投与されている患者に対して、本剤を150mgで投与することに変更された。本剤の承認された用量は1回150mgである。表1)国際共同第III相無作為化比較試験(MONARCH2試験)における成績 本剤+フルベストラント
投与群プラセボ+フルベストラント
投与群症例数 (日本人症例数)
446(64)
223(31)
イベント発現例数
222
157
無増悪生存期間中央値(月)
(95%信頼区間)16.4
(14.4-19.3)
9.3
(7.4-11.4)
ハザード比
(95%信頼区間)
0.553
(0.449-0.681)層別ログランク検定(両側)
p<0.0000001
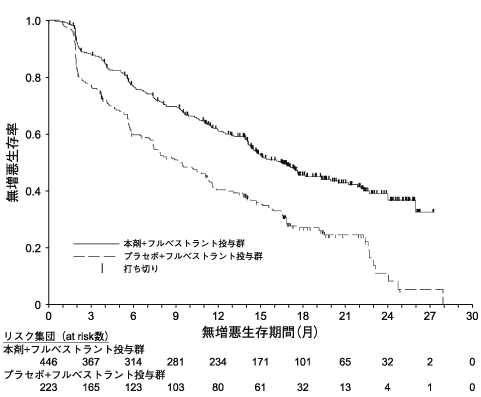
図1)無増悪生存期間のKaplan-Meier曲線(MONARCH2試験)本剤が投与された441例(日本人63例を含む)に認められた主な副作用は、下痢(80.0%)、好中球減少症(41.6%)、悪心(34.4%)、疲労(30.9%)、腹痛(27.5%)等であった。
-
17.1.2 国際共同第III相無作為化比較試験(MONARCH3試験)
ホルモン受容体陽性かつHER2陰性であり、内分泌治療歴のない手術不能又は再発閉経後乳癌患者493例を対象に、本剤+非ステロイド性アロマターゼ阻害剤(nonsteroidal aromatase inhibitor:NSAI、レトロゾール又はアナストロゾール)とプラセボ+NSAIを比較する無作為化二重盲検プラセボ対照第III相国際共同試験を実施した。
本剤150mg又はプラセボ(1日2回)とレトロゾール2.5mg又はアナストロゾール1mg(1日1回)を病態の悪化等が認められるまで投与を継続した。
主要評価項目である治験責任医師判定による無増悪生存期間について、本剤+NSAIの併用投与により、プラセボ+NSAIの併用投与と比較して統計学的に有意な延長が認められた13) 。
表2)国際共同第III相無作為化比較試験(MONARCH3試験)における成績 本剤+NSAI投与群
プラセボ+NSAI投与群
症例数 (日本人症例数)
328(38)
165(15)
イベント発現例数
108
86
無増悪生存期間中央値(月)
(95%信頼区間)NE
14.73
(11.11-17.46)ハザード比
(95%信頼区間)0.543
(0.409-0.723)層別ログランク検定(両側)
p=0.000021
NE:推定不能
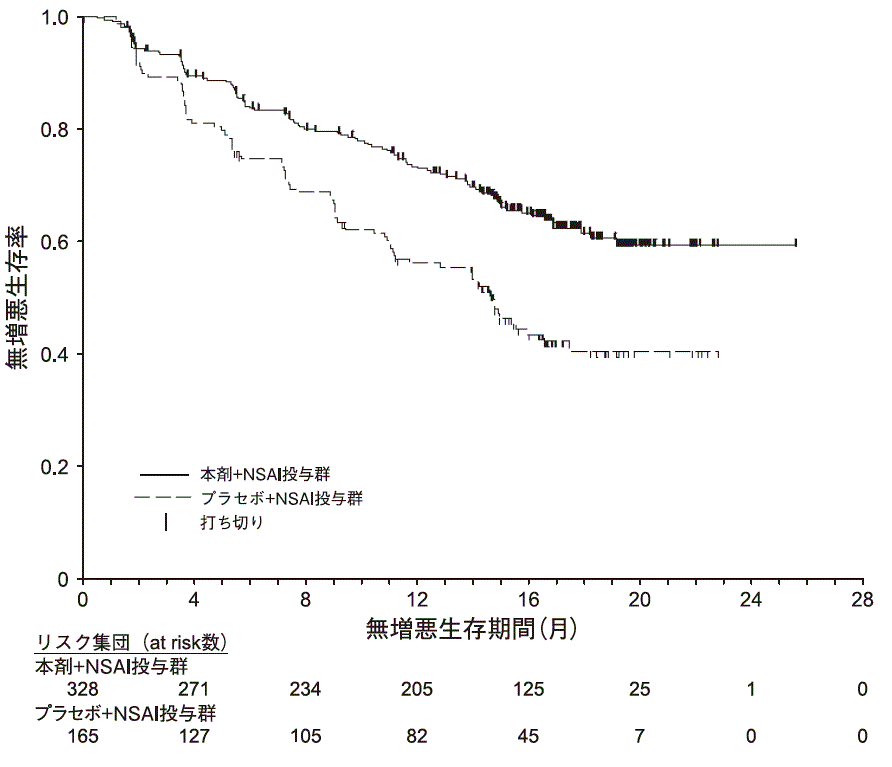
図2)無増悪生存期間のKaplan-Meier曲線(MONARCH3試験)本剤が投与された327例(日本人38例を含む)に認められた主な副作用は、下痢(78.9%)、好中球減少症(39.1%)、疲労(31.8%)、悪心(30.3%)、貧血(24.2%)等であった。
-
17.1.3 国際共同第III相無作為化比較試験(monarchE試験)
ホルモン受容体陽性かつHER2陰性の乳癌の術後患者 注12) 5637例を対象に、本剤+内分泌療法と内分泌療法を比較する無作為化非盲検第III相国際共同試験を実施した。
本剤150mg(1日2回)は病態の悪化等が認められるまで最長24ヵ月間投与し、医師選択の標準的な内分泌療法剤(タモキシフェン、アロマターゼ阻害剤等の単独投与、又はLH-RHアゴニストとの併用投与)は5~10年間投与することとされ、再発又は投与中止基準に該当するまで継続することとされた。
主要評価項目である治験責任医師判定による浸潤性疾患のない生存期間について、本剤+内分泌療法の併用投与(2808例、うち日本人181例)により、内分泌療法(2829例、うち日本人196例)と比較して統計学的に有意な延長が認められた(ハザード比(95%信頼区間):0.747(0.598-0.932)、p=0.00957(層別ログランク検定)、有意水準(両側)0.0264)14) 。(2020年3月16日データカットオフ)
再発高リスク 注13) の術後乳癌患者における浸潤性疾患のない生存期間は下表のとおりであった15) 。,
注12) 登録された患者のうち、化学療法歴及び放射線療法歴のある患者はそれぞれ94.4%及び95.4%であった。注13) 再発高リスクの定義は、以下①又は②のいずれかの基準(monarchE試験コホート1の患者選択基準に相当)に該当することとする。
①病理検査で同側腋窩リンパ節の4個以上で転移陽性。
②病理検査で同側腋窩リンパ節の1~3個で転移陽性(術前薬物療法前の細胞診も可)、かつ原発腫瘍径5cm以上(術前薬物療法前の画像検査も可)又はModified Bloom-Richardson grading systemによる組織学的グレード316) 。表3)国際共同第III相無作為化比較試験における再発高リスクの術後乳癌患者(monarchE試験コホート1)の成績 本剤+内分泌療法群
内分泌療法群
症例数 (日本人症例数)
2555(169)
2565(175)
イベント発現例数
127
182
2年無浸潤疾患生存率
(95%信頼区間)
92.1
(90.3-93.6)
88.4
(86.2-90.2)
ハザード比
(95%信頼区間)
0.714
(0.569-0.896)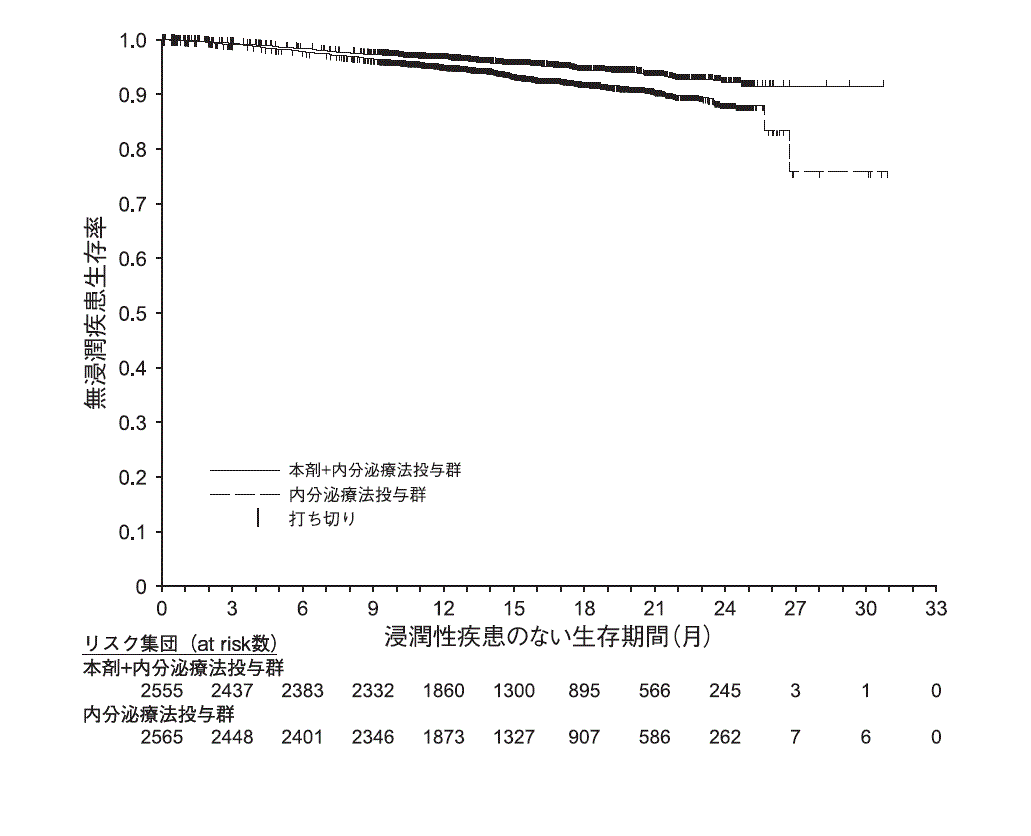
図3)再発高リスクの術後乳癌患者における浸潤性疾患のない生存期間のKaplan-Meier曲線(monarchE試験コホート1)本剤が投与された2791例(日本人181例を含む)に認められた主な副作用は、下痢(79.1%)、好中球減少症(42.6%)、白血球減少症(34.4%)、疲労(29.2%)、腹痛(27.6%)等であった。
18. 薬効薬理
19. 有効成分に関する理化学的知見
一般的名称
アベマシクリブ(Abemaciclib)〔JAN〕
化学名
N-{5-[(4-Ethylpiperazin-1-yl)methyl]pyridin-2-yl}-5-fluoro-4-[4-fluoro-2-methyl-1-(1-methylethyl)-1H-benzimidazol-6-yl]pyrimidin-2-amine
分子式
C27H32F2N8
分子量
506.59
性状
白色~黄色の粉末である。
化学構造式
![]()
融点
約176℃
21. 承認条件
医薬品リスク管理計画を策定の上、適切に実施すること。
23. 主要文献
1) Fujiwara Y, et al.: Cancer Chemother Pharmacol. 2016; 78(2): 281-288
2) 社内資料: 進行癌患者を対象としたアベマシクリブの外国第I相試験(JPBA試験)(2018年9月21日承認、CTD2.7.6.6)
3) 社内資料: アベマシクリブの絶対的バイオアベイラビリティ(JPBS試験)(2018年9月21日承認、CTD2.7.6.2)
4) 社内資料: アベマシクリブの薬物動態に及ぼす食事の影響(JPCC試験)(2018年9月21日承認、CTD2.7.6.3)
5) 社内資料: アベマシクリブ及び代謝物のヒト血漿蛋白結合率(2018年9月21日承認、CTD2.7.2.2.1.1)
6) 社内資料: In vitroにおける代謝及び薬物相互作用の検討(2018年9月21日承認、CTD2.7.2.2.1.3)
7) 社内資料: アベマシクリブのマスバランス試験(JPBD試験)(2018年9月21日承認、CTD2.7.6.13)
8) 社内資料: 様々な重症度の肝機能障害を有する被験者におけるアベマシクリブの薬物動態(JPBV試験)(2018年9月21日承認、CTD2.7.6.9)
9) 社内資料: アベマシクリブとクラリスロマイシンの相互作用(JPBE試験)(2018年9月21日承認、CTD2.7.6.10)
10) 社内資料: アベマシクリブとリファンピシンの相互作用(JPBF試験)(2018年9月21日承認、CTD2.7.6.11)
11) 社内資料: アベマシクリブとメトホルミンの相互作用(JPCK試験)(2018年9月21日承認、CTD2.7.6.12)
12) Sledge GW, et al.: J. Clin. Oncol. 2017; 35(25): 2875-2884
13) Goetz MP, et al.: J. Clin. Oncol. 2017; 35(32): 3638-3646
14) *Johnston SRD, et al.: J. Clin. Oncol. 2020; 38(34): 3987-3998
15) *社内資料: 早期乳癌患者を対象としたアベマシクリブの無作為化非盲検第III相国際共同試験(monarchE試験)(2021年12月24日承認、審査報告書)
16) *Elston CW, Ellis IO.: Histopathology. 1991; 19(5): 403-410
17) Gelbert LM, et al.: Invest. New Drugs. 2014; 32(5): 825-837
18) Torres-Guzman R, et al.: Oncotarget. 2017; 8(41): 69493-69507
24. 文献請求先及び問い合わせ先
日本イーライリリー株式会社 医薬情報問合せ窓口
〒651-0086 神戸市中央区磯上通5丁目1番28号
TEL:0120-360-605(医療関係者向け)
www.lillymedical.jp
26. 製造販売業者等
26.1 製造販売元
日本イーライリリー株式会社
神戸市中央区磯上通5丁目1番28号
Ⓡ:登録商標