アブラキサン点滴静注用100mg
作成又は改訂年月
| * | 2021年 8月改訂 ( 第1版 、 用法変更 ) |
日本標準商品分類番号
薬効分類名
承認等
一般的名称
3. 組成・性状
5. 効能又は効果に関連する注意
6. 用法及び用量
*乳癌にはA法又はE法を、胃癌にはA法又はD法を、非小細胞肺癌にはB法を、治癒切除不能な膵癌にはC法を使用する。
A法:
通常、成人にはパクリタキセルとして、1日1回260mg/m2(体表面積)を30分かけて点滴静注し、少なくとも20日間休薬する。これを1コースとして、投与を繰り返す。
なお、患者の状態により適宜減量する。
B法:
通常、成人にはパクリタキセルとして、1日1回100mg/m2(体表面積)を30分かけて点滴静注し、少なくとも6日間休薬する。週1回投与を3週間連続し、これを1コースとして、投与を繰り返す。
なお、患者の状態により適宜減量する。
C法:
ゲムシタビンとの併用において、通常、成人にはパクリタキセルとして、1日1回125mg/m2(体表面積)を30分かけて点滴静注し、少なくとも6日間休薬する。週1回投与を3週間連続し、4週目は休薬する。これを1コースとして、投与を繰り返す。
なお、患者の状態により適宜減量する。
D法:
通常、成人にはパクリタキセルとして、1日1回100mg/m2(体表面積)を30分かけて点滴静注し、少なくとも6日間休薬する。週1回投与を3週間連続し、4週目は休薬する。これを1コースとして、投与を繰り返す。
なお、患者の状態により適宜減量する。
E法:
他の抗悪性腫瘍剤との併用において、通常、成人にはパクリタキセルとして、1日1回100mg/m2(体表面積)を30分かけて点滴静注し、少なくとも6日間休薬する。週1回投与を3週間連続し、4週目は休薬する。これを1コースとして、投与を繰り返す。
なお、患者の状態により適宜減量する。
7. 用法及び用量に関連する注意
-
〈効能共通〉
-
7.1 本剤の投与にあたっては下記に留意し、必要に応じ休薬、減量を実施すること。
- A法、B法又はE法
好中球数及び血小板数の変動に十分留意し、次コース投与前の臨床検査で好中球数が1,500/mm3未満又は血小板数が100,000/mm3未満であれば、骨髄機能が回復するまで投与を延期すること。また、B法又はE法の同一コース内の投与にあたっては、投与前の臨床検査で好中球数が500/mm3未満又は血小板数が50,000/mm3未満であれば、骨髄機能が回復するまで投与を延期すること。投与後、好中球数が7日間以上にわたって500/mm3未満となった場合、血小板数が50,000/mm3未満となった場合、又は発熱性好中球減少症が発現した場合、更にB法又はE法では次コース投与開始が7日間以上延期となる好中球減少が発現した場合も次コースの投与量を減量すること。
また、高度(Grade 3)な末梢神経障害が発現した場合には、軽快又は回復(Grade 1以下)するまで投与を延期し、次回の投与量を減量すること。,,,,, - C法
〈第1日目(各コース開始時)〉
好中球数及び血小板数の変動に十分留意し、投与前の臨床検査で好中球数が1,500/mm3未満又は血小板数が100,000/mm3未満であれば、骨髄機能が回復するまで投与を延期すること。
〈第8及び15日目〉第8日目
投与前血液検査(/mm3)
対応
①
好中球数1,000超
かつ
血小板数75,000以上投与量変更なし
②
好中球数500以上1,000以下
又は
血小板数50,000以上75,000未満1段階減量
③
好中球数500未満
又は
血小板数50,000未満休薬
第15日目
投与前血液検査
(/mm3)第8日目での
血液検査の結果対応
好中球数1,000超
かつ
血小板数75,000以上①の場合
投与量変更なし
②の場合
第1日目投与量に増量可
③の場合
1段階減量
好中球数500以上
1,000以下
又は
血小板数50,000以上
75,000未満①の場合
投与量変更なし
②の場合
第8日目投与量に同じ
③の場合
1段階減量
好中球数500未満
又は
血小板数50,000未満①~③の場合
休薬
投与後、好中球数が7日間以上にわたって500/mm3未満となった場合、血小板数が50,000/mm3未満となった場合、又は発熱性好中球減少症が発現した場合には、次回の投与量を減量すること。
また、高度(Grade 3)な末梢神経障害が発現した場合には、軽快又は回復(Grade 1以下)するまで投与を延期し、次回の投与量を減量すること。,,,,, - D法
好中球数及び血小板数の変動に十分留意し、投与前の臨床検査で好中球数が1,000/mm3未満又は血小板数が75,000/mm3未満であれば、骨髄機能が回復するまで投与を延期すること。
本剤と他の抗悪性腫瘍剤との併用の場合は、第1日目の投与前の臨床検査で好中球数が1,500/mm3未満又は血小板数が100,000/mm3未満であれば、骨髄機能が回復するまで投与を延期すること。
投与後、好中球数が500/mm3未満となった場合、血小板数が25,000/mm3未満となった場合、又は発熱性好中球減少症が発現した場合には、次回の投与量を減量すること。
また、高度(Grade 3)な末梢神経障害が発現した場合には、軽快又は回復(Grade 2以下)するまで投与を延期し、次回の投与量を減量すること。,,,,, - 減量の目安
減量段階
A法
B法又はE法
C法
D法
通常投与量
260mg/m2
100mg/m2
125mg/m2
100mg/m2
1段階減量
220mg/m2
75mg/m2
100mg/m2
80mg/m2
2段階減量
180mg/m2
50mg/m2
75mg/m2
60mg/m2
- A法、B法又はE法
-
7.1 本剤の投与にあたっては下記に留意し、必要に応じ休薬、減量を実施すること。
- 〈非小細胞肺癌及び乳癌〉
- 〈胃癌〉
8. 重要な基本的注意
- 8.1 本剤の使用にあたっては、下記を患者に説明し、理解を得るよう努めること。
- 8.2 本剤の添加物である人血清アルブミンの原料となる血漿については、HBs抗原、抗HCV抗体、抗HIV-1抗体及び抗HIV-2抗体が陰性であることを確認している。さらに、プールした試験血漿については、HBV-DNA、HCV-RNA及びHIV-1-RNAについて核酸増幅検査(NAT)を実施し、適合した血漿を本剤の製造に使用しているが、当該NATの検出限界以下のウイルスが混入している可能性が常に存在する。人血清アルブミンの製造工程である、Cohn低温エタノール分画法及び60±0.5℃ 10~11時間の液状加熱処理は、HIVをはじめとする各種ウイルスに対し、除去・不活化効果を有することが確認されているが、本剤投与による感染症発生の可能性は否定できないので、投与後の経過を十分に観察すること。
- 8.3 添加物に使用している人血清アルブミンの現在の製造工程では、ヒトパルボウイルスB19などのウイルスを完全に不活化・除去することが困難であるため、本剤の投与によりその感染の可能性を否定できないので、投与後の経過を十分に観察すること。
- 8.4 現在までに本剤の投与により変異型クロイツフェルト・ヤコブ病(vCJD)などが伝播したとの報告はない。しかしながら、本剤の添加物である人血清アルブミンの製造工程において異常プリオンを低減し得るとの報告があるものの、理論的なvCJDなどの伝播のリスクを完全には排除できないので、本剤投与の際には患者への説明を十分行い、治療上の必要性を十分検討の上投与すること。
- 8.5 骨髄抑制などの重篤な副作用が起こることがあるので、頻回に臨床検査(血液検査、肝機能検査、腎機能検査等)を行うこと。また、使用が長期間にわたると副作用が強くあらわれ、遷延性に推移することがあるので、投与は慎重に行うこと。なお、白血球減少が軽度であっても著明な好中球減少を発現する症例を認めていることから、血液検査の際には、白血球分画の測定を実施すること。また、本剤の投与にあたってはG-CSF製剤の適切な使用に関しても考慮すること。,,,
- 8.6 末梢神経障害が高頻度に起こるので、患者の状態を十分に観察すること。使用が長期間にわたると発現頻度が高くなる傾向にあるので、投与は慎重に行うこと。,
- 8.7 重篤な過敏反応が起こることがあるので、観察を十分に行い、重篤な過敏症状(呼吸困難、胸痛、低血圧、頻脈、徐脈、潮紅、血管浮腫、発汗等)があらわれた場合には、直ちに投与を中止し、適切な処置を行うこと。本剤投与中は頻回にバイタルサイン(血圧、脈拍数)のモニタリングを行うなど、患者の状態を十分に観察すること。
- 8.8 低血圧、高血圧、徐脈等が起こることがあるので、本剤投与中は頻回にバイタルサイン(血圧、脈拍数)のモニタリングを行うなど、患者の状態を十分に観察すること。重篤な刺激伝導障害があらわれた場合には、適切な処置を行い、その後の本剤投与に際しては継続的に心電図のモニタリングを行うなど、患者の状態を十分に観察すること。
- 8.9 関節痛及び筋肉痛が高頻度に起こるので、観察を十分に行い、症状があらわれた場合には鎮痛剤投与等の適切な処置を行うこと。
- 8.10 発熱が起こることがあるので、観察を十分に行い、症状があらわれた場合には感染に対する管理を十分に行い、解熱剤投与等の適切な処置を行うこと。
- 8.11 投与初期又は比較的低用量の投与でも副作用があらわれることがあるので、使用上の注意に十分注意すること。
- 8.12 出血傾向の発現又は増悪に十分注意すること。
9. 特定の背景を有する患者に関する注意
10. 相互作用
10.2 併用注意(併用に注意すること)
| 薬剤名等 | 臨床症状・措置方法 | 機序・危険因子 |
|---|---|---|
|
放射線照射 |
パクリタキセルに胸部への放射線照射を併用した場合に、重篤な食道炎又は肺臓炎が発現したとの報告がある。併用する場合には、患者の状態に注意し、食道炎や肺陰影等が出現した場合には、本剤の投与及び放射線照射を直ちに中止し、適切な処置を行うこと。 |
機序は不明であるが、動物試験(マウス)でパクリタキセルによる放射線感受性増加が認められている。 |
|
放射線照射 |
骨髄抑制等を増強することがあるので、併用する場合には、患者の状態を観察しながら、本剤を減量するか又は投与間隔を延長すること。 |
骨髄抑制等の予想される副作用項目が重複している。 |
|
抗悪性腫瘍剤 |
骨髄抑制等の副作用が増強するおそれがある。併用療法を行う場合には、患者の状態を観察しながら、減量するか又は投与間隔を延長すること。 |
骨髄抑制等の予想される副作用が重複している。 |
|
シスプラチン |
パクリタキセルをシスプラチンの後に投与した場合、逆の順序で投与した場合より骨髄抑制が増強するおそれがある。併用療法を行う場合には、本剤をシスプラチンの前に投与すること。 |
パクリタキセルをシスプラチンの後に投与した場合、パクリタキセルのクリアランスが低下し、パクリタキセルの血中濃度が上昇する。 |
|
シスプラチン |
末梢神経障害が増強するおそれがある。併用療法を行う場合には、患者の状態を観察しながら、減量するか又は投与間隔を延長すること。 |
末梢神経障害が予想される副作用として重複している。 |
|
ドキソルビシン塩酸塩 |
パクリタキセルをドキソルビシンの前に投与した場合、逆の順序で投与した場合より骨髄抑制が増強するおそれがある。併用療法を行う場合には、本剤をドキソルビシンの後に投与すること。 |
パクリタキセルをドキソルビシンの前に投与した場合、ドキソルビシンのクリアランスが低下し、ドキソルビシンの血中濃度が上昇する。 |
|
ドキソルビシン塩酸塩 |
心毒性が増強するおそれがある。併用療法を行う場合には、患者の状態を観察しながら、減量するか又は投与間隔を延長すること。 |
胆汁排泄の競合により、ドキソルビシン及びその代謝物であるドキソルビシノールの血中濃度が上昇する。 |
|
ビタミンA、アゾール系抗真菌剤(ミコナゾール等)、マクロライド系抗生剤(エリスロマイシン等)、ステロイド系ホルモン剤(エチニルエストラジオール等)、ジヒドロピリジン系カルシウムチャンネルブロッカー(ニフェジピン等)、シクロスポリン、ベラパミル塩酸塩、キニジン硫酸塩水和物、ミダゾラム、ラパチニブトシル酸塩水和物 |
骨髄抑制等の副作用が増強するおそれがある。併用療法を行う場合には、患者の状態を観察しながら、減量するか又は投与間隔を延長すること。 |
併用薬剤がCYP2C8、CYP3A4等を阻害し、パクリタキセルの代謝が阻害され、パクリタキセルの血中濃度が上昇する。 |
11. 副作用
11.1 重大な副作用
-
11.1.1 *白血球減少などの骨髄抑制
好中球減少(51.9%)、白血球減少(29.6%)、リンパ球減少(6.1%)、貧血[ヘモグロビン減少(31.4%)、ヘマトクリット値減少(1.1%)、赤血球減少(1.1%)等]、血小板減少(17.7%)、汎血球減少(0.3%)等があらわれることがある。また、骨髄抑制の持続により、発熱性好中球減少症(2.9%)等の感染症の併発が報告されている。,,,
-
11.1.2 *感染症
好中球減少の有無にかかわらず敗血症(0.8%)等の感染症があらわれ、死亡に至る例が報告されている。異常が認められた場合には、抗菌薬の投与等の適切な処置を行うこと。
- 11.1.3 *末梢神経障害(60.8%)、麻痺(頻度不明)
-
11.1.4 脳神経麻痺(0.1%未満)
顔面神経麻痺、声帯麻痺等の脳神経麻痺があらわれることがある。
-
11.1.5 *ショック(頻度不明)、アナフィラキシー(0.4%)
呼吸困難、胸痛、低血圧、頻脈、徐脈、潮紅、血管浮腫、発汗等の異常が認められた場合には投与を中止し、適切な処置を行うこと。
-
11.1.6 *間質性肺疾患(1.6%)
発熱、咳嗽、呼吸困難及び胸部X線検査異常等が認められた場合には投与を中止し、副腎皮質ホルモン剤の投与等の適切な処置を行うこと。
-
11.1.7 急性呼吸窮迫症候群(0.1%未満)
急速に進行する呼吸困難、低酸素症、両側性びまん性肺浸潤影等の胸部X線異常等が認められた場合には投与を中止し、適切な処置を行うこと。
- 11.1.8 *心筋梗塞(0.2%)、うっ血性心不全(0.4%)、心伝導障害(0.1%未満)
- 11.1.9 脳卒中(0.1%未満)、肺塞栓(0.2%)、肺水腫(0.1%)、血栓性静脈炎(0.2%)
- 11.1.10 難聴(0.1%未満)、耳鳴(0.3%)
- 11.1.11 消化管壊死(頻度不明)、消化管穿孔(頻度不明)、消化管出血(0.6%)、消化管潰瘍(0.3%)
-
11.1.12 *重篤な腸炎(0.6%)
出血性大腸炎、偽膜性大腸炎、虚血性大腸炎等があらわれることがあるので、激しい腹痛・下痢等があらわれた場合には投与を中止し、適切な処置を行うこと。
-
11.1.13 腸管閉塞(0.2%)、腸管麻痺(頻度不明)
腸管閉塞、腸管麻痺(食欲不振、悪心・嘔吐、著しい便秘、腹痛、腹部膨満あるいは腹部弛緩及び腸内容物のうっ滞等)を来し、麻痺性イレウスに移行することがあるので、腸管閉塞、腸管麻痺があらわれた場合には投与を中止し、腸管減圧法等の適切な処置を行うこと。
- 11.1.14 *肝機能障害(1.3%)、黄疸(0.1%)
-
11.1.15 膵炎(0.1%未満)
血清アミラーゼ値等に異常が認められた場合には投与を中止するなど適切な処置を行うこと。
-
11.1.16 *急性腎障害(0.3%)
BUN、血清クレアチニン、クレアチニン・クリアランス値等に異常が認められた場合には投与を中止するなど適切な処置を行うこと。
- 11.1.17 中毒性表皮壊死融解症(Toxic Epidermal Necrolysis:TEN)(頻度不明)、皮膚粘膜眼症候群(Stevens-Johnson症候群)(頻度不明)
-
11.1.18 播種性血管内凝固症候群(DIC)(頻度不明)
血小板数、血清FDP値、血漿フィブリノゲン濃度等の血液検査に異常が認められた場合には投与を中止し、適切な処置を行うこと。
11.2 その他の副作用
|
20%以上 |
5~20%未満 |
5%未満 |
頻度不明 |
|
|---|---|---|---|---|
|
*皮膚及び皮下組織障害 |
脱毛(症)(64.8%)、発疹 |
そう痒症、爪の異常 |
顔面腫脹、蕁麻疹、手足症候群、皮膚乾燥、色素沈着、光線過敏症 |
強皮症様変化 |
|
神経系障害 |
味覚異常 |
嗜眠、めまい、頭痛、運動失調、振戦、反射減弱、注意力障害 | ||
|
*全身障害及び投与局所様態 |
倦怠感(36.7%) |
無力症、発熱、浮腫 |
疼痛、胸痛、注射部位反応、悪寒 | |
|
*胃腸障害 |
悪心(31.9%)、下痢 |
口内炎、嘔吐、便秘 |
腹痛、消化不良、腹部膨満(感)、口内乾燥、嚥下障害、口唇炎、舌痛 | |
|
筋骨格系及び結合組織障害 |
関節痛、筋肉痛 |
四肢痛、骨痛、背部痛、胸壁痛、筋力低下、筋痙縮 | ||
|
代謝及び栄養障害 |
食欲不振 |
脱水(症) | ||
|
*臨床検査 |
ALT上昇、AST上昇 |
γ-GTP上昇、Al-P上昇、クレアチニン上昇、カリウム上昇、カリウム低下、ビリルビン上昇、アルブミン減少、カルシウム低下、ナトリウム低下、好酸球数増多、総蛋白減少、血糖値上昇、尿糖陽性、尿蛋白陽性、体重減少 | ||
|
呼吸器、胸郭及び縦隔障害 |
鼻出血 |
呼吸困難、咽喉頭痛、咳嗽、胸水、鼻炎、喀血、発声障害、しゃっくり | ||
|
眼障害 |
視力異常、眼痛、眼乾燥、角膜炎、結膜炎、流涙、黄斑浮腫 | |||
|
精神障害 |
不眠症、不安、うつ病 | |||
|
血管障害 |
高血圧、潮紅、低血圧 | |||
|
腎及び尿路障害 |
尿失禁 | |||
|
心臓障害 |
頻脈、不整脈、徐脈 | |||
|
耳及び迷路障害 |
耳痛 | |||
|
生殖系及び乳房障害 |
乳房痛 |
14. 適用上の注意
14.1 薬剤調製時の注意
- 14.1.1 懸濁液の調製に当たっては、必ず生理食塩液を使用すること。また、本懸濁液は他の薬剤とは混注しないこと。
- 14.1.2 本剤は細胞毒性を有するため、調製時には手袋を着用することが望ましい。皮膚に本剤又は懸濁液が付着した場合は、直ちに多量の流水及び石けんでよく洗い流すこと。
- 14.1.3 懸濁液は調製後速やかに使用するか、又は箱に戻し、冷蔵庫(2~8℃)に遮光保存して8時間以内に使用すること。
- 14.1.4 点滴バッグ中に入れた懸濁液は速やかに使用すること。
- 14.1.5 使用前に懸濁液に未懸濁物、沈殿物が認められ、再懸濁させても沈殿物が認められた場合は使用しないこと。
- 14.1.6 調製時に、注射針に塗布されているシリコーン油により不溶物を生じることがある。調製後に懸濁液中に不溶物がないか目視で確認すること。不溶物が認められた場合は使用しないこと。
14.2 懸濁液調製方法
- 14.2.1 無菌的環境下にて、患者の体表面積にあわせ必要なバイアルを準備し、アルコールでゴム栓を拭う。
- 14.2.2 1バイアル当たり生理食塩液20mLをバイアルの内壁伝いに、直接、内容物にかけないよう泡立ちに注意しながらゆっくりと注入する。(この操作は、泡立ちの発生を最小限にするため重要である。)
- 14.2.3 内容物が確実に濡れるよう5分間以上バイアルを静置する。
- 14.2.4 内容物が十分に濡れたら、均一な白色ないし黄色の懸濁液になるまで、静かに円弧を描くように回したり、緩やかに上下に転倒を繰り返して混和する。(泡立ちに注意する。)
- 14.2.5 調製した懸濁液は必要量をバイアルから抜き取り、事前に用意した空の点滴バッグ等にゆっくりと注入する。
注意:懸濁液を生理食塩液に入れて希釈しないこと。
15. その他の注意
16. 薬物動態
16.1 血中濃度
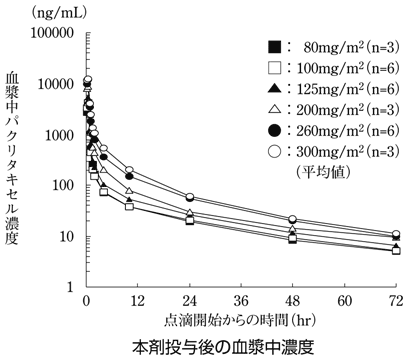
16.3 分布
ラットに本剤の3H標識体を投与した24時間後における組織内放射能濃度は、脳を除く各臓器・組織で高く、速やかに移行した。また、前立腺、肝臓、肺、精嚢、膵臓、脾臓、消化管、腎臓で血液・血漿より高かった。120時間後における放射能が高かった組織は肝臓、肺、精巣及び卵巣であった2) 。
16.4 代謝
16.5 排泄
外国人乳癌患者に本剤260mg/m2を30分間点滴静注したときの未変化体パクリタキセルの尿中排泄量の平均値は約4%であり、これは腎外での消失が主な排泄経路であることを示している。代謝物である6α-ヒドロキシパクリタキセル及び3´-p-ヒドロキシパクリタキセルの尿中排泄率は総投与量の1%以下であった。糞中には総投与量の約20%が排泄された4) 。
16.6 特定の背景を有する患者
-
16.6.1 肝機能障害患者
AST及びビリルビンに基づいて肝機能障害の程度を分類し、その障害の程度に応じ3用量(130mg/m2、200mg/m2、260mg/m2) 注2) を設定し、本剤の薬物動態について検討した5) 。
肝機能障害の程度
投与量
(n)AUCinf
(ng・hr/mL)CL
(L/hr/m2)AST
かつ
ビリルビン
>ULN-
<10×ULN>ULN-
≦1.25×ULN260mg/m2
(5)11983±4335
23.8±7.5
1.26-2.0×ULN
200mg/m2
(4)8660±2891
24.9±7.0
2.01-5.0×ULN
130mg/m2
(5)7146±1326
18.7±3.1
ULN:基準値上限。AUCinf及びCLは平均±標準偏差を示した。
AST≧10×ULNあるいはビリルビン>5.0×ULNの肝機能障害患者については検討されていない。注2) 本剤の承認用量はA法:260mg/m2、B法、D法及びE法:100mg/m2、C法:125mg/m2である。
17. 臨床成績
17.1 有効性及び安全性に関する試験
-
〈胃癌〉
-
17.1.1 国内第Ⅲ相試験
フッ化ピリミジン系抗悪性腫瘍剤を含む初回化学治療に不応となった胃癌患者を対象として、本剤(A群(A法):3週を1コースとして、本剤260mg/m2を1日目に投与、及びB群(D法):4週を1コースとして、本剤100mg/m2を週1回(1、8及び15日目)投与し、4週目(22日目)を休薬)と、他のパクリタキセル製剤(C群:4週を1コースとして、他のパクリタキセル製剤80mg/m2を週1回(1、8及び15日目)投与し、4週目(22日目)を休薬)を比較した試験結果は以下のとおりであった6) 。
群
(例数)A群(A法)
(243)B群(D法)
(240)C群
(243)生存期間中央値(月)
10.3
11.1
10.9
ハザード比a
(信頼区間)1.06
(0.87~1.31)b0.97
(0.76~1.23)c-
p値d
0.062
0.0085
-
a:多変量Cox比例ハザードモデル。
b:C群に対するA群のハザード比及び95%信頼区間。95%信頼区間の上限値が非劣性マージンとして設定した1.25を上回り、非劣性は示されなかった。
c:C群に対するB群のハザード比及び97.5%信頼区間。97.5%信頼区間の上限値が非劣性マージンとして設定した1.25を下回り、非劣性が示された。
d:非劣性の仮説検定に対応する片側p値。A法の副作用の発現率は99.6%(243/244例)であり、主な副作用は末梢神経障害(84.8%)、好中球減少(81.6%)、脱毛症(80.7%)、白血球減少(63.9%)、食欲減退(38.5%)、関節痛(38.5%)、筋肉痛(35.2%)であった。D法の副作用の発現率は98.8%(238/241例)であり、主な副作用は脱毛症(82.6%)、末梢神経障害(66.0%)、好中球減少(65.6%)、白血球減少(56.8%)であった。
-
17.1.2 国内第Ⅱ相試験
フッ化ピリミジン系抗悪性腫瘍剤を含む初回化学治療に不応となった胃癌患者を対象とした試験(本剤260mg/m2 30分点滴静注3週ごと投与)における奏効率は、27.8%(15/54例)であった7) 。
副作用の発現率は100%(55/55例)であり、主な副作用は脱毛(94.5%)、末梢神経障害(92.7%)、白血球減少(85.5%)、好中球減少(78.2%)、関節痛(65.5%)、筋肉痛(63.6%)、発疹(54.5%)、食欲不振(52.7%)、貧血(38.2%)、リンパ球減少(38.2%)、悪心(38.2%)、ALT上昇(36.4%)、AST上昇(34.5%)、口内炎(32.7%)であった。 -
17.1.3 国内第Ⅱ相試験
フッ化ピリミジン系抗悪性腫瘍剤を含む初回化学治療に不応となった胃癌患者を対象に本剤とラムシルマブを併用投与した試験(4週を1コースとして、本剤100mg/m2を1、8及び15日目に、ラムシルマブ8mg/kgを1及び15日目に投与し、4週目(22日目)を休薬)における奏効率は、54.8%(23/42例)であった8) 。,
副作用の発現率は100%(43/43例)であり、主な副作用は脱毛(93.0%)、好中球減少(90.7%)、末梢神経障害(58.1%)、鼻出血(46.5%)、高血圧(41.9%)、白血球減少(37.2%)であった。
-
17.1.1 国内第Ⅲ相試験
-
〈乳癌〉
-
17.1.4 海外第Ⅲ相試験
乳癌患者を対象に実施した本剤と他のパクリタキセル製剤との試験(本剤260mg/m2 30分点滴静注3週ごと投与)における奏効率は、24.0%(55/229例)であった4) 。
本剤
(260mg/m2)
(30分点滴静注、3週ごと投与)他のパクリタキセル製剤
(175mg/m2)
(3時間点滴静注、3週ごと投与)症例数(例)
229
225
奏効例(例)
55
25
標的病変奏効率(%)
24.0
11.1
95%信頼区間(%)
18.48~29.55
7.00~15.22
本剤における副作用の発現率は99.1%(227/229例)であり、主な副作用は脱毛(90.4%)、末梢神経障害(70.7%)、関節痛(31.9%)、好中球減少(33.6%)であった。
-
17.1.5 国際共同第Ⅲ相試験(IMpassion130試験)
化学療法歴のないホルモン受容体陰性かつHER2陰性の手術不能又は再発乳癌患者の初回治療において、本剤とアテゾリズマブの併用投与(A群:4週を1コースとして、本剤100mg/m2を1、8及び15日目に、アテゾリズマブ840mgを1及び15日目に投与し、4週目(22日目)を休薬)と、本剤とプラセボの併用投与(B群:4週を1コースとして、本剤100mg/m2を1、8及び15日目に、プラセボを1及び15日目に投与し、4週目(22日目)を休薬)を比較した試験について、全体集団のうち、PD-L1陽性集団(日本人25例を含む)の結果は以下のとおりであった9) 。
群
(例数)PD-L1陽性集団
ハザード比
(信頼区間)p値
A群
(185)B群
(184)無増悪生存期間
中央値(月)7.46
4.96
0.62
(0.49~0.78)<0.0001a
a:層別log-rank検定
A群(日本人34例を含む)における副作用の発現率は96.5%(436/452例)であり、主な副作用は、脱毛(56.2%)、末梢神経障害(52.2%)、倦怠感(43.4%)、悪心(41.2%)、好中球減少(33.2%)であった。また、主な免疫関連副作用は甲状腺機能障害(甲状腺機能低下症(12.6%)、甲状腺機能亢進症(3.8%)等)、副腎機能障害(副腎機能不全(0.4%)等)であった。
-
17.1.6 *国際共同第Ⅲ相試験(KEYNOTE-355試験)
化学療法歴のないホルモン受容体陰性かつHER2陰性の手術不能又は再発乳癌患者の初回治療において、本剤を含む化学療法 注3) とペムブロリズマブ 注4) の併用投与(A群)と、本剤を含む化学療法 注3) とプラセボ 注5) の併用投与(B群)を比較した試験について、全体集団のうち、PD-L1陽性(CPS 注6) ≧10)集団(日本人28例を含む)の結果は以下のとおりであった10) 。
群
(例数)PD-L1陽性
(CPS≧10)集団ハザード比a
(信頼区間)p値b
A群
(220)B群
(103)無増悪生存期間
中央値(月)9.7
5.6
0.65
(0.49~0.86)0.0012
a:層別Cox比例ハザードモデルによるプラセボ+化学療法との比較
b:層別log-rank検定A群のうち本剤とペムブロリズマブの併用投与(日本人5例を含む)における副作用の発現率は95.3%(164/172例)であり、主な副作用は脱毛(51.7%)、貧血及び悪心(各30.8%)、好中球減少(29.1%)、下痢(26.7%)、疲労(23.8%)であった。また、主な免疫関連副作用は甲状腺機能障害(甲状腺機能低下症(17.4%)、甲状腺機能亢進症(4.7%)、甲状腺炎(1.7%)等)であった。
注3) 以下より担当医師が患者ごとに選択。
本剤100mg/m2(4週を1コースとして、各コースの1、8、15日目に投与)、ゲムシタビン1,000mg/m2及びカルボプラチンAUC 2mg・min/mL相当量(3週を1コースとして、各コースの1、8日目に投与)又はパクリタキセル90mg/m2(4週を1コースとして、各コースの1、8、15日目に投与)。注4) 200mgを3週間間隔で投与。注5) 3週間間隔で投与。注6) PD-L1を発現した細胞数(腫瘍細胞、マクロファージ及びリンパ球)を総腫瘍細胞数で除し、100を乗じた値。
-
17.1.4 海外第Ⅲ相試験
-
〈非小細胞肺癌〉
-
17.1.7 国際共同第Ⅲ相試験
非小細胞肺癌患者(Stage ⅢB/Ⅳ)の初回治療において、本剤とカルボプラチンの併用投与(A群:3週を1コースとして、1日目に本剤100mg/m2及びカルボプラチンAUC=6 注7) を投与し、8日目と15日目に本剤100mg/m2を投与)と、他のパクリタキセル製剤とカルボプラチンの併用投与(T群:3週を1コースとして、1日目に他のパクリタキセル製剤200mg/m2及びカルボプラチンAUC=6を投与)を比較した試験結果は以下のとおりであった11) 。
A群
T群
p値
奏効率(例数)
33%(170/521)
25%(132/531)
0.005a
a:カイ二乗検定
A群(日本人72例を含む)における副作用の発現率は91.2%(469/514例)であり、主な副作用は好中球減少(59.1%)、脱毛(55.8%)、貧血(48.8%)、末梢神経障害(45.5%)、血小板減少(44.7%)であった。
注7) カルボプラチンの投与量は、カルバートの式「投与量(mg/body)=AUC目標値×[GFR(糸球体濾過率)+25]」に従って算出した。
-
17.1.7 国際共同第Ⅲ相試験
-
〈治癒切除不能な膵癌〉
-
17.1.8 海外第Ⅲ相試験
遠隔転移を有する膵癌患者の初回治療において、本剤とゲムシタビンの併用投与(A群:4週を1コースとして、本剤125mg/m2及びゲムシタビン1,000mg/m2を週1回(1、8及び15日目)投与し、4週目(22日目)を休薬)と、ゲムシタビン単独投与(B群:4週を1コースとして、ゲムシタビン1,000mg/m2を週1回(1、8及び15日目)投与し、4週目(22日目)を休薬(第1サイクルのみ22日目にも投与)) 注8) を比較した試験結果は以下のとおりであった12) 。
群
(例数)A群
(431)B群
(430)ハザード比
p値
生存期間中央値(月)
8.5
6.7
0.72
<0.001a
a:層別log-rank検定
A群における副作用の発現率は95.7%(403/421例)であり、主な副作用は疲労226例(53.7%)、脱毛211例(50.1%)、悪心207例(49.2%)、末梢神経障害206例(48.9%)、貧血194例(46.1%)、好中球減少193例(45.8%)、下痢156例(37.1%)、血小板減少149例(35.4%)、末梢性浮腫141例(33.5%)、嘔吐133例(31.6%)であった。
注8) 膵癌に対するゲムシタビンの承認用法及び用量は、ゲムシタビンとして1,000mg/m2を週1回3週投与し、1週間休薬である。
-
17.1.8 海外第Ⅲ相試験
18. 薬効薬理
19. 有効成分に関する理化学的知見
一般的名称
パクリタキセル(Paclitaxel)
化学名
(-)-(1S,2S,3R,4S,5R,7S,8S,10R,13S)-4,10-Diacetoxy-2-benzoyloxy-5,20-epoxy-1,7-dihydroxy-9-oxotax-11-en-13-yl(2R,3S)-3-benzoylamino-2-hydroxy-3-phenylpropionate
分子式
C47H51NO14
分子量
853.91
化学構造式

20. 取扱い上の注意
22. 包装
1バイアル
23. 主要文献
1) *臨床第Ⅰ相試験におけるABI-007投与後の薬物動態データを用いた用量比例性の解析(承認年月日:2010年7月23日、CTD2.7.2.2、2.7.2.5)
2) Sparreboom, A., et al. : Clin. Cancer Res. 2005 ; 11(11): 4136-4143
3) Rochat, B. : Clin. Pharmacokinet. 2005 ; 44(4): 349-366
6) Shitara, K., et al. : Lancet Gastroenterol, Hepatol. 2017 ; 2(4): 277-287
7) Sasaki, Y., et al. : Cancer Sci. 2014 ; 105(7): 812-817
8) Bando, H., et al.:Eur. J. Cancer. 2018 ; 91 : 86-91
9) Schmid, P., et al.:N. Engl. J. Med. 2018 ; 379(22): 2108-2121
10) Cortes, J., et al. : Lancet. 2020 ; 396(10265): 1817-1828
11) Socinski, M.A., et al. : J. Clin. Oncol. 2012 ; 30(17): 2055-2062
12) Von Hoff, D.D., et al. : N. Engl. J. Med. 2013 ; 369(18): 1691-1703
13) Schiff, P.B., et al. : Nature. 1979 ; 277(5698): 665-667
14) Schiff, P.B., et al. : Proc. Natl. Acad. Sci. USA. 1980 ; 77(3): 1561-1565
15) Desai, N., et al. : Clin. Cancer Res. 2006 ; 12(4): 1317-1324
16) Awasthi, N., et al. : Carcinogenesis. 2013 ; 34(10): 2361-2369
24. 文献請求先及び問い合わせ先
大鵬薬品工業株式会社 医薬品情報課
〒101-8444 東京都千代田区神田錦町1-27
TEL 0120-20-4527
26. 製造販売業者等
26.1 製造販売元
大鵬薬品工業株式会社
東京都千代田区神田錦町1-27
26.2 提携先
Abraxis BioScience
米国